Part II of the series: The future of flexible manufacturing
When designing a complex biopharma manufacturing facility, the list of potential risks is seemingly endless. How to tackle this? Achieve true manufacturing flexibility, support your business driver, and satisfy regulatory authorities with a focus on three key risk drivers.
During the conceptual design (CD) phase of a new project, what are some of the key risk elements that must be addressed to ensure that true flexibility between products, customers and batches can be achieved within both regulatory and business guidelines? It is a long list, so in the interest of space, let’s discuss three:
- Validation of closure
- Segregation strategy
- Change-over
Validation of closure. When is closed, closed?
A challenge that many companies face is settling how to define and validate closure. This is especially true when single-use systems (SUS) are the primary process platform. For this discussion, we will focus on SUS closure validation, as it often presents the biggest challenge today.
There are multiple definitions and approaches to a closed system, but here we will take the BioPhorum Operations Group (BPOG) three-step process for executing this type of risk analysis by asking three questions:
- Do you understand the process?
- What is the process state and requirements?
- Is the process adequately closed?
A risk assessment of this kind should consider the closure scale, bioburden control requirement and/or process requirement. For example, a proven tool that can easily be implemented to assess risk is the structured Failure Mode and Effects Analysis (FMEA) approach.[i]
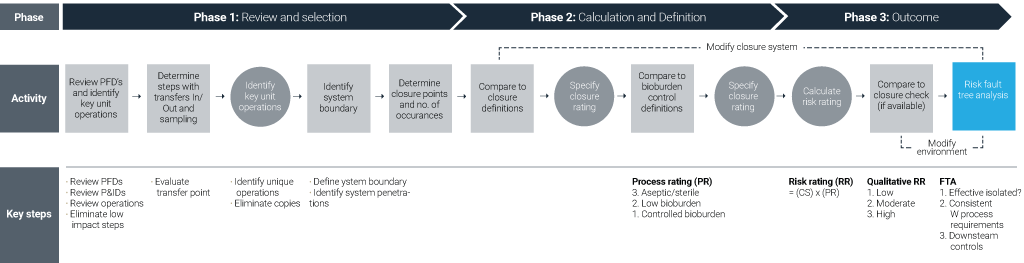
Figure 1. Systematic Process Closure Analysis (BPOG).
The goal of carrying out this risk assessment is to clearly define where closure risk is at its greatest and develop engineering solutions to effectively mitigate that risk to meet regulatory needs and expectations.
The industry trend towards closed processes allows manufacturers to rely less on environmental control. With this trend comes a growing movement to operate in a Controlled Non-Classified (CNC) space, both in new facilities and existing facilities where a risk-based analysis of system closure has been completed and validated. The drivers for switching to CNC spaces include:
- Product protection: Engineering controls safer than environmental controls (cleanroom)
- Inspection performance: Avoids answering “20 questions” from third parties
- Capital expenditure (CAPEX) reduction: fewer, smaller cleanrooms, equipment in shared CNC space
- Operating expense (OPEX) reduction: cleanroom HVAC, testing, monitoring, gowning and manning
Based on the closure analysis, one of the next critical aspects to support the segregation strategy is to assign an area classification based on bioburden contamination risk. The output from this risk assessment provides a recommended area classification, using a logical approach for impact evaluation, risk mitigation and detection. This results in a standard risk score where a recommended area classification can be derived.
Segregation strategy
Segregation strategy should be aligned with the overall control strategy of the manufacturing operation. Separation of products and processes is established and validated conceptually by using different combinations of the enterprise elements referenced from the three sides of the enterprise triangle.[ii] Elements of all three are required for any effective segregation strategy.
The capabilities of the facility layout should support:
- High throughput, multi-product manufacturing around SUS or hybrid platforms by efficiently managing the various flows of components, product and waste
- Flow schemes that can be both bidirectional and unidirectional
- Entrance and extraction of equipment and components within the manufacturing space on a just-in-time basis to maximize operational efficiency
In the third article of this series, “Maximize efficiency: Minimize operational interference in Biomanufacturing,” we identify two layout options that require a different focus on segregation strategy. Based on segregation strategy analysis, the strengths and weaknesses of each option are shown below.[iii]
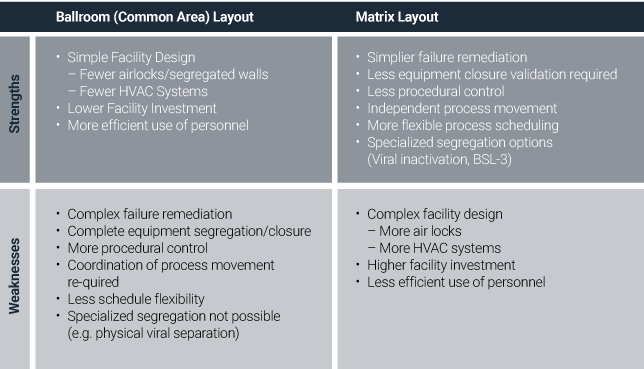
One important area of segregation focus is around viral clearance. Here, process closure and viral inactivation approach play key roles in the segregation strategy. While physical segregation is the primary means taken by many companies, other methods of segregation include:
- Laminar flow utilized in combination with layout "geography" as in ballroom configurations
- Enhanced gowning controls
- Improvement to primary containment via systems such as isolators
Change-over
The US Food and Drug Administration (FDA) defines change-over as, “A logical series of steps performed to assure that the multi-use processing suite(s) and equipment have been properly cleared before processing a different product.”[iv] The regulatory assessment focus includes:
- New product/process impact
- Product characterisation
- Cross-contamination risk
- System closure
- Cleaning validation
- Containment
Developing a truly flexible manufacturing platform layout is much like a well-choreographed dance. The assessment of all manufacturing “steps” requires a well-defined time-and-motion analysis. During the CD phase of the project, this will require a clear identification and understanding of the project knowns and unknowns.
Typical knowns
- Customer
- Project location
- Project phasing cycle
- End date
- Production scale
- Block flow diagrams
Typical unknowns
- Exact process/product
- Process material balance
- Multi-product/phase approach
- Capital budget allocation
- Customer project team
- Final equipment vendors
- Biosafety considerations
- Staff capabilities or roles
- Storage requirements
- User requirements
- Ergonomic and Health Safety Environment preferences
- Raw materials
From this comes the development and execution of a "step-by-step" choreography of activities.

Figure 3[v]. A step-by-step choreography of activities.
Such an analysis is critical to ensure that change-over can be performed in the most efficient manner, which allows for the complete production schedule of multi-product platforms to support the business need and ensure regulatory issues are addressed. Flexibility and efficiency are improved when the change-over process is mapped in such a manner, analyzing the steps, and eliminating, resequencing, or improving standardization of activities.
This analysis will identify critical layout attributes such as space requirements for staging and transfer, access/egress/transfer requirements, required connections counts and storage. These will be translated into specific layouts and flows to support the multi-product options.
The key to change-over efficiency rests in the ability to validate the multi-use aspect of the operation. Know in advance which products will be produced and their Critical Quality Attributes (CQA) and Critical Process Parameters (CPPs) based on the identified process. Ensure that specific assays are in place for the active components. Finally, make sure, as part of the infrastructure of the enterprise, that adequate change control procedures have been implemented. Losses in facility flexibility will occur when time, personnel and material waste are consumed by product change-over.[vi]
[i] “Closed Systems: Why is Closure Analysis So Important?” Jeff Odum, CPIP, Global Technology Partner
[ii] “The Key Design Drivers for Developing Flexible Manufacturing Assets” Jeff Odum, CPIP, Global Technology Partner
[iii]“Impact of Facility Layout on Developing and Validating Segregation Strategies in the Next Generation of Multi-product, Multi-phase Biopharmaceutical Manufacturing Facilities” Mark Witcher, PhD. Pharmaceutical Engineering Supplement, 2015.
[iv] “FDA Expectations: Multi-product Facility Considerations for Biological Products”, Lori Peters, FDA/CBER, ISBiotech Conference March 2013.
[v] “Managing the Design of Single-use Facilities”, Jeff Odum, CPIP, ISPE Mid-West Forum, 2016
[vi] Handbook of Industrial Biotechnology, Wiley & Sons, 2007




